
Endocrinology
Treatment Updates
 |
This activity has been planned and implemented in
accordance with Essentials and Standards of the Accreditation Council for
Continuing Medical Education (ACCME) through the joint sponsorship of
Medical Education Collaborative and Medscape. Medical Education
Collaborative, a non-profit education organization, is accredited by the
ACCME to provide continuing medical education for physicians and takes
responsibility for the content, quality and scientific integrity of this
CME activity.
This activity has also been planned and implemented
in accordance with the Quality Criteria of the American Council on
Pharmaceutical Education (ACPE) through the cosponsorship of Medical Education Collaborative,
Inc. and Medscape.
CME in this activity indicates continuing
education for medical professionals. Please click here
for eligibility requirements.
This activity is made possible by an
unrestricted educational grant(s) provided to Medical Education
Collaborative by Novartis.
 |
Mealtime Glucose Excursions and Glycemic Control in Type 2
Diabetes CME
Edward Horton, MD
http://www.medscape.com/Medscape/Endocrinology/TreatmentUpdate/2000/tu01/public/toc-tu01.html
Valid for CME until January 7, 2001
If you cannot register online and complete the activity,
you may also receive credit by mailing your completed Registration form, Post
Test and Evaluation Forms to:
 |
Medical Education Collaborative
1800 Jackson
Street, Suite 200
Golden, CO 80401
Telephone: (303) 278-1900
|
Goal
The goal of this Treatment Update is to provide an overview of
the pathogenesis of type 2 diabetes and how postprandial hyperglycemia affects
overall glycemic control. An overview of therapeutic strategies that target
physiologic mealtime insulin requirements will be presented.
Learning Objectives
Mealtime Glucose Excursions and Glycemic Control
in Type 2 Diabetes is intended for physicians and pharmacists.
Upon completion of this self-study activity, participants will be able to:
- Outline the major metabolic defects that contribute to hyperglycemia in
type 2 diabetes
- Describe the insulin secretory response to a meal in type 2 diabetes
- Explain how postprandial hyperglycemia contributes to overall glycemic
control
- Discuss the rationale for tailoring treatment to mealtime glucose
excursions
- Review strategies that reduce postprandial glucose excursions and, as a
result, improve overall glycemic control
Eligibility for Credit
Continuing education credit will be awarded
to US physicians, physician assistants, and pharmacists who
successfully complete this activity as described in the section Instructions
for Credit. For all other medical professionals who successfully complete this
activity, Medscape will issue a Letter of Completion. For information on
applicability and acceptance of continuing education credit for this activity,
please consult your professional licensing boards.
Instructions for Credit
Participation in this self-study activity
should be completed in approximately one (1) hour. There are no fees for
participating and receiving CME credit for this activity. To successfully
complete this activity and receive credit, participants must follow these
steps during the period January 7, 2000 through January 7,
2001.
- Register for continuing education credit by completing the
"registration" process.
- Read the learning objectives.
- Read the article text and tables, and review figures.
- Read, complete, and submit answers to the post test questions and
evaluation questions. Participants must receive a test score of at least
70%, and respond to all evaluation questions to receive certificate by mail.
Certificates will be electronically mailed to all eligible participants upon
successful completion of the post test and submission of the activity
evaluation for this activity. Certificates will be mailed to all eligible
participants within 4-6 weeks of completion of this activity to those
participants who are unable to print the certificate or who submit the post
test and evaluation form for this activity using regular mail.
Legal Disclaimer
The
material presented here does not reflect the views of Medscape, Medical Education Collaborative and the
companies providing unrestricted educational grants or the authors and writers.
These materials may discuss uses and dosages for therapeutic products that have
not been approved by the United States Food and Drug Administration. All readers
and continuing education participants should verify all information and consult
a qualified health care professional before treating patients or utilizing any
therapeutic product discussed in this continuing education activity.
Table of Contents
- Abstract
- Introduction
- Etiology of Hyperglycemia
- First-phase Insulin Secretion and Normal Glucose Tolerance
- Mealtime Glucose Excursions Contribute to Overall Poor Glycemic Control
in Type 2 Diabetes
- Postprandial Hyperglycemia and the Complications of Diabetes
- Improving Mealtime Glucose Control by Restoring Early Insulin Secretion
in Type 2 Diabetes
- Summary
Abstract
Type 2 diabetes mellitus is characterized by a transition from
normal glucose tolerance, to impaired glucose tolerance, to overt diabetes.
There is convincing evidence that the risk of developing microvascular
complications of diabetes, including retinopathy, nephropathy, and neuropathy,
is related to the degree of hyperglycemia. Type 2 diabetes is also associated
with an increased morbidity and mortality from macrovascular disease, including
coronary artery disease, peripheral vascular disease, and stroke. Postprandial
hyperglycemia contributes substantially to overall glycemic control, and in
general, its role in the pathogenesis of the long-term microvascular and
macrovascular complications of diabetes has been underestimated. It has been
recognized that neither sulfonylureas nor regular insulin can create the
appropriately brief increases in insulin secretion that normally occur in
response to meals. Therefore, pharmacologic agents with a shorter onset and
duration of action have been developed to more closely approximate the normal
insulin response to meals to reduce postprandial hyperglycemia. This approach
offers patients the ability to establish tight control of blood glucose levels
and allows patients to be more flexible with their day-to-day calorie intake.
This treatment update will review strategies that reduce postprandial glucose
excursions and as a result, improve overall glycemic control.
Introduction
Type 2 diabetes mellitus is associated with substantial
morbidity and mortality from microvascular and macrovascular complications. The
risk of developing diabetic microvascular complications is related to the degree
of hyperglycemia, with the risk increasing at values only slightly above the
normal range for hemoglobin A1c (HbA1c) and progressively increasing as
hyperglycemia worsens.[1] There does not appear
to be a threshold glucose level for microvascular complications that is
significantly above the accepted upper limits of normal for fasting and
postprandial glucose concentrations.[2-4]
Based on the results of the Diabetes Control and Complications Trial
(DCCT)[4,5] in patients with type 1 diabetes, as
well as the Kumomoto Study [6] and the United
Kingdom Prospective Diabetes Study (UKPDS)[7] in
patients with type 2 diabetes, it is generally accepted that intensive treatment
that achieves normal blood glucose levels minimizes the risk of developing
long-term complications of diabetes. Improving glycemic control can prevent
microvascular complications of both type 1[4]
and type 2 diabetes.[8] Although the
relationship between macrovascular disease and glycemic control is less clear
than it is for microvascular disease, hyperglycemia is a significant risk factor
for developing macrovascular disease in type 2 diabetes.[2,9,10] In the UKPDS, there was a nonsignificant 16%
reduction in risk of myocardial infarction in the intensively treated group
compared with those treated by conventional modalities.[7]
Etiology of Hyperglycemia
In recent years, our understanding of the
pathophysiology of type 2 diabetes has increased dramatically. As illustrated in
Figure 1, there are 3 major metabolic defects that contribute to hyperglycemia
in type 2 diabetes.[11,12]
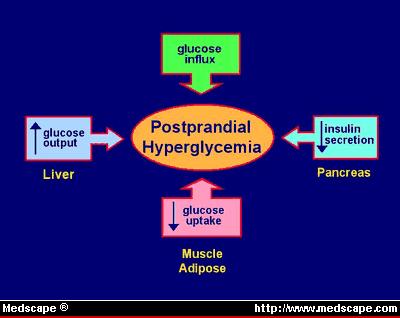
Figure 1. Metabolic defects of type 2 diabetes.
First, insulin-stimulated glucose uptake in
insulin-sensitive tissues, particularly skeletal muscle and adipose tissue, is
decreased as a result of insulin resistance. Second, there is increased hepatic
glucose output in the fasting state and decreased suppression of hepatic glucose
output following meals. This phenomenon is thought to be a manifestation of
insulin resistance in the liver and is a major contributor to both fasting and
postprandial hyperglycemia. Third, a defect in beta-cell function results in
abnormal insulin secretion in response to a glucose stimulus. It appears that
insulin resistance and decreased beta-cell response to glucose occur early in
the pathogenesis of diabetes.[13,14]
In nondiabetic individuals, the beta-cell response to an insulin secretagogue
is biphasic, with an early burst of insulin release occurring during the first
10 minutes after a stimulus, followed by a second-phase insulin secretion that
is characterized by a sustained increase in insulin release that may last
several hours.[15] Whereas the early event
appears to represent the release of preformed insulin, the later phase may
reflect the release of newly synthesized insulin in response to continued
beta-cell stimulation. An early defect in the development of diabetes is the
loss of the early or first-phase insulin secretion following food ingestion or
glucose administration.[13,16,17] This results
in a delayed and late-peaking plasma insulin profile, which is associated with
decreased glucose tolerance. As type 2 diabetes progresses, there is a gradual
deterioration of beta-cell function that results in a loss of both the early and
later phases of insulin secretion.[18]
During the early stages of type 2 diabetes, the transition from normal to
impaired glucose tolerance and finally to overt type 2 diabetes is primarily
associated with episodes of postprandial hyperglycemia.[19] Once diabetes is established, there is a
progressive worsening of hyperglycemia in both the fasting and the postprandial
state.
First-phase Insulin Secretion and Normal Glucose Tolerance
The
importance of early or first-phase insulin secretion in maintaining normal
glucose tolerance has been demonstrated in a number of studies. Calles-Escadon
and Robbins[20] reported that a 20-minute
infusion of somatostatin inhibited first-phase insulin secretion in normal
volunteers and resulted in a conversion from normal to impaired glucose
tolerance. In a study by Bruttomesso and colleagues,[21] the administration of lispro insulin to patients
with type 2 diabetes undergoing an oral glucose tolerance test produced a rapid
increase in insulin concentration, which peaked much earlier and disappeared
much faster compared with regular insulin. This resulted in a significant
improvement in glucose tolerance. Furthermore, because the insulin concentration
rapidly decreased, it more closely simulated a normal physiologic response to a
glucose challenge than was achieved by regular insulin. A more physiologic
insulin response would be expected to decrease the risk for hypoglycemia during
the late postprandial period and also avoid chronic hyperinsulinemia, which may
be associated with weight gain, insulin resistance,[22] lipid abnormalities,[23] and hypertension[24] ; all are potential cardiovascular risk factors.
Mealtime Glucose Excursions Contribute to Overall Poor Glycemic Control in
Type 2 Diabetes
Frequently, guidelines and/or recommendations advise
monitoring fasting plasma glucose (FPG) and HbA1c concentrations to evaluate
overall glycemic control. However, FPG does not provide information about the
contribution of the postprandial rise in glucose levels to overall glycemic
control, and HbA1c does not provide information about the daily oscillations in
blood glucose levels because it only represents the average glucose levels
during the previous 2 to 3 months. Because patients are most often in a
postprandial state rather than in a truly fasting state, and wide fluctuations
in plasma glucose (PG) levels may occur throughout the day -- with high values 1
to 2 hours after a meal and low values before the next meal -- it may be more
useful to assess postprandial glucose levels to monitor overall glycemic
control. In addition, postprandial glucose levels may more closely represent the
metabolic processes involved in the pathogenesis of type 2 diabetes -- insulin
resistance, increased hepatic glucose output, and impaired insulin secretion.
A recent study by Avignon and colleagues,[25]
evaluated the relative importance of measuring PG at different time points in
assessing glucose control in patients with type 2 diabetes. A total of 66 adult
patients with type 2 diabetes treated with diet or diet plus oral medications
were enrolled in the study. Multiple daily blood samples were taken at 8:00 AM
(fasting), 11:00 AM (1 hour before lunch), 2:00 PM (2 hours after the beginning
of lunch), and 5:00 PM (5 hours after the beginning of lunch). Patients were
then divided into 3 groups according to their HbA1c concentrations: less than
7%, 7% to 8.5%, and greater than 8.5%. Correlations between the various blood
glucose determinations and overall glycemic control as measured by HbA1c were
carried out. Figure 2 shows that the mean glycemic profile corresponds to the
level of glycemic control.
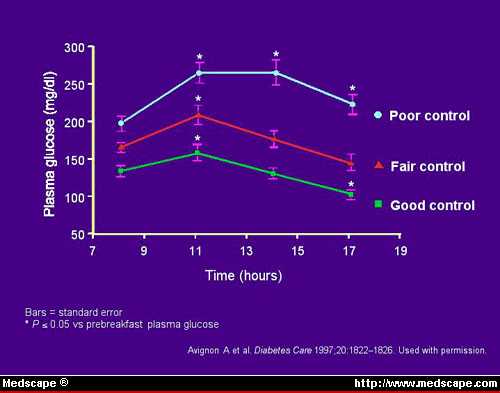
Figure 2. Mean glycemic profile relative to
glycemic control.
Prebreakfast PG levels had the weakest correlation, and postlunch PG levels
showed the strongest. Multiple linear regression analysis of the data showed
that postlunch PG and extended postlunch PG levels correlated significantly and
independently with HbA1c values, whereas prebreakfast PG and prelunch PG levels
did not (Figure 3).

Figure 3. Correlation between plasma glucose and
HbA1c.
The results of this study indicate that good glycemic control (HbA1c
concentration less than 7%) is characterized by extended postlunch PG levels
that are lower than the fasting value, whereas poor glycemic control is
associated with an extended postlunch PG levels higher than fasting. In other
words, FPG was not a good predictor of blood glucose concentrations at other
times of the day. The best correlation between PG and HbA1c was evident with the
2:00 PM and 5:00 PM samples, which represent the early and extended postlunch
values. This study illustrates that postprandial increases in PG levels are
better predictors of overall glycemic control than FPG and that both the early
and sustained increases in PG following a meal make a significant contribution
to overall glycemic control.
Postprandial Hyperglycemia and the Complications of Diabetes
Because
postprandial glucose excursions contribute to the overall level of glycemic
control, they are clearly related to the development of the microvascular and
macrovascular complication of type 2 diabetes.
The level of postprandial hyperglycemia is associated with an increased risk
of developing cardiovascular disease.[26-30]
Along with meal-related glucose excursions, type 2 diabetes is also associated
with a postprandial increase in lipids,[31-34]
particularly very low-density lipoproteins and the activation of a prothrombotic
state.[35,36]
In the majority of studies demonstrating an association between glycemia and
risk for cardiovascular disease, fasting glucose or HbA1c concentrations have
been used as static parameters for measuring overall glycemic control. However,
in studies such as the Paris Prospective Study[37,38] and the Honolulu Heart Study,[39] increased cardiovascular risk has been seen in
individuals with either impaired glucose tolerance or with increasing glucose
concentrations that fall within the normal range. Because these latter groups
predominantly experience postprandial increases in blood glucose levels,
postprandial hyperglycemia may play an important role in determining the risk
for macrovascular disease.
Several studies have shown a better correlation between 2-hour PG
concentration and the risk of cardiovascular disease than with FPG.[28,40] Furthermore, the degree of risk, which is
conferred by the 2-hour PG concentration, is almost twice that associated with
HbA1c.[28] Hanefeld and colleagues[30] examined the relationship between fasting glucose
and postprandial glucose concentration with respect to myocardial infarction and
mortality during an 11-year period in patients with type 2 diabetes. They found
that although there was no significant relationship between fasting blood
glucose levels and cardiovascular risk factors, there was a significant
relationship between cardiovascular risk and postprandial glucose concentration.
All of this suggests that the postprandial increases in glucose concentration
may produce physiologic effects that are not reflected by fasting glucose or
HbA1c. Unfortunately, the assessment of fluctuations in blood glucose levels
throughout the day requires frequent and invasive blood glucose testing. The
measurement of 1,5-anhydro-D-glucitol has been proposed as one way to assess
glucose fluctuations throughout the day, but this is not seen as a practical
application.[41] The recent approval of the
GlucoWatch automatic glucose biographer (Cygnus Inc, Redwood City, Calif), a
device that makes frequent, noninvasive measurements of glucose possible, may
facilitate intensive management of diabetes.[42]
In summary, the goal of treatment of patients with type 2 diabetes should be
to achieve the best possible overall glycemic control by restoring a normal,
physiologic insulin response to feeding and decreasing late postprandial insulin
levels and chronic hyperinsulinemia. However, this is difficult to achieve using
currently available medications because excessive postprandial increases in PG
and hypoglycemia during the late postprandial or fasting state are major
obstacles to achieving a completely normal blood glucose profile. Consequently,
the American Diabetes Association established the target HbA1c concentration at
less than 7% or as close to normal as possible (4%-6%) and recommended a need
for changing the therapeutic regimen if the HbA1c concentration is higher than
8%.[43]
Improving Mealtime Glucose Control by Restoring Early Insulin Secretion in
Type 2 Diabetes
Whereas sulfonylureas or insulin may adequately control
fasting blood glucose levels, it is usually more challenging to modulate
postprandial glucose excursions. Because neither sulfonylureas nor regular
insulin appropriately simulate the postprandial insulin response to a meal, they
are not ideal for managing postprandial hyperglycemia. Furthermore, because
these agents produce increases in insulin levels that extend beyond the
postprandial period, they may predispose the patient to hypoglycemia. The
rationale for tailoring pharmacologic therapy to mealtimes is based on the
importance of restoring the mealtime insulin secretion profiles of patients with
type 2 diabetes to reestablish the tight control of blood glucose levels during
the postprandial period. Furthermore, this approach also takes into account that
most people with type 2 diabetes are overweight and are advised to reduce
calorie consumption, but the risk of hypoglycemia does not allow them to be
flexible about their day-to-day calorie intake.[44]
Dietary modifications may somewhat diminish postprandial hyperglycemia, but
they often do not produce a satisfactory response, and therefore pharmacologic
intervention is usually undertaken. Several classes of pharmacologic agents are
either currently available or in clinical development for managing postprandial
glucose excursions. These include alpha-glucosidase inhibitors, short-acting
insulinotropic agents, rapid-acting insulin analogues, and amylin analogues.
Alpha-glucosidase Inhibitors
Alpha-glucosidase inhibitors, such as
acarbose, miglitol, and voglibose, delay the digestion of complex carbohydrates
by competitively inhibiting intestinal alpha-glucosidases that hydrolyze
oligosaccharides into monosaccharides. By delaying the digestion and prolonging
the intestinal absorption of dietary carbohydrates, these agents diminish
postprandial hyperglycemia. However, because the total amount of carbohydrate
absorbed is not reduced, there are no net energy losses.[45,46]
Clinical studies have shown that alpha-glucosidase inhibitors reduce
postprandial glucose levels and improve overall glycemic control as indicated by
HbA1c.[47-52] In addition, some studies suggest
that acarbose reduces the postprandial increase in triglycerides[49] and may have beneficial effects on
lipoproteins.[50] Compared with sulfonylureas,
they are not associated with postprandial hypoglycemia.[51] In a comparative study of miglitol and acarbose,
both agents produced comparable reductions in HbA1c concentrations.[53] When used as monotherapy, these agents may not
achieve overall glycemic control in all patients, especially in those with
elevated FPG levels.
Patients should be instructed to take their dose of alpha-glucosidase
inhibitor with the first bite of each meal. The principal side effects of
alpha-glucosidase inhibitors are related to their effect on the gastrointestinal
tract and are primarily manifested as flatulence and loose stools. They do not
appear to affect the rate of gastric emptying.[54] Tolerance to these side effects usually occurs
with continued administration.
Short-acting Insulinotropic Agents
Because these agents more rapidly
stimulate insulin secretion compared with sulfonylureas, they simulate a more
physiologic increase in mealtime insulin levels. Thus, they are primarily used
for reducing postprandial hyperglycemia.
Repaglinide, a carbamoylmethyl benzoic acid derivative structurally related
to meglitinide,[55] is the first short-acting
insulinotropic agent approved in the United States for the treatment of type 2
diabetes. Repaglinide augments glucose-stimulated insulin secretion by closing
ATP-sensitive potassium [K+(ATP)] channels on beta cells.[55] This causes depolarization of the beta cell and
the opening of voltage-sensitive calcium channels allowing the influx of
extracellular calcium ions, which in turn, stimulates insulin release.
Repaglinide more effectively increases insulin release from islet cells
incubated in vitro in the presence of D-glucose or other nutrients than in their
absence.[55, 56]
Repaglinide significantly increases postprandial insulin levels and also
decreases mean FPG, postprandial PG levels, and HbA1c.[57,58] It is more effective than glipizide and
similar to glibeclamide and gliclazide in maintaining overall glycemic control
as measured by HbA1c.[59] Patients should be
instructed to take repaglinide within 15 minutes of a meal, but this time may
vary from immediately preceding the meal to as long as 30 minutes before.
Patients who skip a meal should be instructed to skip the dose for that meal.
Conversely, if a meal is added, then patients should be instructed to add a
dose.
Nateglinide, another short-acting insulinotropic agent, is an amino acid
derivative that stimulates insulin secretion from pancreatic beta cells by
closing K+(ATP) channels.[60] In vitro studies
using rat cardiac myocytes and vascular smooth muscle cells suggest that
nateglinide is more selective for beta-cell K+(ATP) channels than repaglinide
and glyburide.[60] Nateglinide blunts mealtime
glucose excursions, returning glucose levels to predose values 4 hours after a
meal.[61]
Rapid-acting Insulin Analogues
The tendency of human insulin to
self-associate under normal physiologic conditions results in a slow and
prolonged absorption from the subcutaneous site of injection. This requires that
regular human insulin be injected approximately 30 to 60 minutes before a
meal,[62] as administration immediately before a
meal produces less than optimal insulin levels during the early phase of glucose
absorption and hyperinsulinemia by the time meal absorption is complete.
Structural modifications of the insulin molecule have produced insulin analogues
that have a weaker tendency to self-associate and, as a result, are more rapidly
and reliably absorbed from the injection site. The short-acting analogues of
insulin, insulin lispro and insulin aspart, were developed to provide a more
physiologic insulin response to food intake.[63]
These insulin analogues are usually administered within 10 to 20 minutes of a
meal, allowing more flexibility in insulin dosing.
Clinical trials in patients with both type 1 and type 2 diabetes have shown
that insulin lispro reduces postprandial glucose excursions.[64] A large, multinational study compared the effect
of lispro injected immediately before a meal with regular human insulin, which
was injected 30 to 45 minutes before eating. Postprandial glucose levels were
lower in patients in the insulin lispro group.[65] When the early rise in plasma insulin was
restored by the lispro analogue, postprandial glucose levels were reduced and
subsequent hyperglycemia and hyperinsulinemia were prevented after an oral
glucose load was compared with regular human insulin.[21] This improvement in glucose tolerance was
associated with a prompter, short-lived suppression of endogenous glucose
production.
The degree of self-association of insulin aspart is similar to that of
insulin lispro. Insulin aspart is absorbed twice as fast and it produces maximum
insulin leves that are approximately twice as high compared with human
insulin.[66] In patients with type 1 diabetes,
insulin aspart significantly improved postprandial blood glucose control after
lunch and dinner and significantly reduced the occurrence of hypoglycemic
episodes requiring third-party intervention.[67]
Amylin Analogues
Amylin is a pancreatic hormone produced by the beta
cell and cosecreted with insulin in response to various secretagogues. Amylin
delays gastric emptying and inhibits postprandial glucagon secretion. In
individuals with type 2 diabetes, the decline in amylin secretory response
correlates with reduced pancreatic beta-cell activity.[68] However, the short half-life limits the use of
amylin. These limitations have been overcome by the advent of pramlintide, an
analogue of amylin.
Recent clinical studies in patients with type 1 diabetes indicate that
pramlintide produces a significant decrease in mean PG and postprandial glucose
concentration despite comparable insulin level.[69] In patients with type 2 diabetes, pramlintide
produced a decrease in HbA1c.[70] However, the
clinical usefulness of pramlintide in type 2 diabetes remains to be defined.
Summary
An early defect in the development of diabetes is the loss of
the early or first-phase insulin response to meals. This results in a delayed
and late-peaking plasma insulin profile, which is associated with decreased
glucose tolerance. Increasing evidence suggests that controlling postprandial
hyperglycemia improves glycemic control, and it is well known that achieving
this slows or prevents the development of diabetic complications. The rationale
for the development of rapid-acting agents that reduce postprandial glucose
excursions is based on providing a more physiologic insulin response to meals.
This approach provides patients with type 2 diabetes the ability to more easily
establish tight glycemic control and offers them the flexibility of modulating
calorie intake without the increased risk of hypoglycemia.
References
- The Diabetes Control and Complications Trial Group. The absence of a
glycemic threshold for the development of long-term complications: the
perspective of the Diabetes Control and Complications Trial. Diabetes.
1996;45:1289-1298.
- Haffner SM. Epidemiological studies on the effects of hyperglycemia and
improvement of glycemic control on macrovascular events in type 2 diabetes.
Diabetes Care. 1999;22:C54-C56.
- Hanssen KF. Blood glucose control and microvascular and macrovascular
complications in diabetes. Diabetes. 1997;46:S101-S103.
- The Diabetes Control and Complications Trial Group. The effect of
intensive treatment of diabetes on the development and progression of
long-term complications in insulin-dependent diabetes mellitus. N Engl J Med.
1993;329:977-986.
- The Diabetes Control and Complications Trial Group. The effect of
intensive diabetes therapy on measures of autonomic nervous system function in
the Diabetes Control and Complications Trial (DCCT). Diabetologia.
1998;41:416-423.
- Ohkubo Y, Kishikawa H, Araki E, et al. Intensive insulin therapy prevents
the progression of diabetic microvascular complications in Japanese patients
with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year
study. Diabetes Res Clin Pract. 1995;28:103-117.
- United Kingdom Prospective Diabetes Study Group. Intensive blood-glucose
control with sulphonylureas or insulin compared with conventional treatment
and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet.
1998;352:837-853.
- Molyneaux LM, Constantino MI, McGill M, Zilkens R, Yue DK. Better
glycaemic control and risk reduction of diabetic complications in Type 2
diabetes: comparison with the DCCT. Diabetes Res Clin Pract. 1998;42:77-83.
- Wild SH, Dunn CJ, McKeigue PM, Comte S. Glycemic control and
cardiovascular disease in type 2 diabetes: a review. Diabetes Metab Res Rev.
1999;15:197-204.
- Wei M, Gaskill SP, Haffner SM, Stern MP. Effects of diabetes and level of
glycemia on all-cause and cardiovascular mortality. The San Antonio Heart
Study. Diabetes Care. 1998;21:1167-1172.
- Ferrannini E. Insulin resistance versus insulin deficiency in
noninsulin-dependent diabetes mellitus: problems and prospects. Endocr Rev.
1998;19:477-490.
- Dinneen SF. The postprandial state: mechanisms of glucose intolerance.
Diabetes Med. 1997;14:S19-S24.
- Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin
secretory dysfunction and insulin resistance in the pathogenesis of type 2
diabetes mellitus. J Clin Invest. 1999;104:787-794.
- Weyer C, Bogardus C, Pratley RE. Metabolic characteristics of individuals
with impaired fasting glucose and/or impaired glucose tolerance. Diabetes.
1999;48:2197-2203.
- Kahn SE, McCulloch DK, Porte D Jr. Insulin secretion in the normal and
diabetic human. In: Alberti KGMM, Zimmet P, DeFronzo RA, Kenn H, eds.
International Textbook of Diabetes Mellitus. 2nd ed. Chichester, England: John
Wiley Sons Ltd; 1997:337-353.
- Swinburn BA, Gianchandani R, Saad MF, Lillioja S. In vivo beta-cell
function at the transition to early non-insulin-dependent diabetes mellitus.
Metabolism. 1995;44:757-764.
- Davies MJ, Rayman G, Grenfell A, Gray IP, Day JL, Hales CN. Loss of the
first phase insulin response to intravenous glucose in subjects with
persistent impaired glucose tolerance. Diabetes Med. 1994;11:432-436.
- Polonsky KS, Given BD, Hirsch LJ, et al. Abnormal patterns of insulin
secretion in non-insulin-dependent diabetes mellitus. N Engl J Med.
1988;318:1231-1239.
- Owens DR, Dolben J, Jones IR, Birtwell J, Luzio SD. Hormonal and glycaemic
responses to serial meals in newly diagnosed non insulin dependent diabetic
patients. Diabetes Metab. 1989;15:1-4.
- Calles-Escandon J, Robbins DC. Loss of early phase of insulin release in
humans impairs glucose tolerance and blunts thermic effect of glucose.
Diabetes. 1987;36:1167-1172.
- Bruttomesso D, Pianta A, Mari A, et al. Restoration of early rise in
plasma insulin levels improves the glucose tolerance of type 2 diabetic
patients. Diabetes. 1999;48:99-105.
- Rizza RA, Mandarino LJ, Genest J, Baker BA, Gerich JE. Production of
insulin resistance by hyperinsulinaemia in man. Diabetologia. 1985;28:70-75.
- Howard BV. Insulin resistance and lipid metabolism. Am J Cardiol.
1999;84:28J-32J.
- Osei K. Insulin resistance and systemic hypertension. Am J Cardiol.
1999;84:33J-36J.
- Avignon A, Radauceanu A, Monnier L. Nonfasting plasma glucose is a better
marker of diabetic control than fasting plasma glucose in type 2 diabetes.
Diabetes Care. 1997;20:1822-1826.
- Tominaga, Eguchi H, Manaka H, Igarashi K, Kato T, Sekikawa A. Impaired
glucose tolerance is a risk factor for cardiovascular disease, but not
impaired fasting glucose: the Funagata Diabetes Study. Diabetes Care.
1999;22:920-924.
- Shaw JE, Hodge AM, de Courten M, Chitson P, Zimmet PZ. Isolated
post-challenge hyperglycaemia confirmed as a risk factor for mortality.
Diabetologia. 1999;42:1050-1054.
- de Vegt F, Dekker JM, Ruhe HG, et al. Hyperglycaemia is associated with
all-cause and cardiovascular mortality in the Hoorn population: the Hoorn
Study. Diabetologia. 1999;42:926-931.
- Barrett-Connor E, Ferrara A. Isolated postchallenge hyperglycemia and the
risk of fatal cardiovascular disease in older women and men: the Rancho
Bernardo Study. Diabetes Care. 1998;21:1236-1239.
- Hanefeld M, Fischer S, Julius U, et al. Risk factors for myocardial
infarction and death in newly detected NIDDM: the Diabetes Intervention Study,
11-year follow-up. Diabetologia. 1996;39:1577-1583.
- Summers LK, Samra JS, Frayn KN. Impaired postprandial tissue regulation of
blood flow in insulin resistance: a determinant of cardiovascular risk?
Atherosclerosis. 1999;147:11-15.
- Mero N, Syvanne M, Taskinen MR. Postprandial lipid metabolism in diabetes.
Atherosclerosis. 1998;(suppl 1):S53-S55.
- Coppack SW. Postprandial lipoproteins in non-insulin-dependent diabetes
mellitus. Diabetes Med. 1997;14:S67-S74.
- De Man FH, Cabezas MC, Van Barlingen HH, Erkelens DW, de Bruin TW.
Triglyceride-rich lipoproteins in non-insulin-dependent diabetes mellitus:
post-prandial metabolism and relation to premature atherosclerosis. Eur J Clin
Invest. 1996;26:89-108.
- Festa A, D'Agostino Jr R, Mykkanen L, et al. Relative contribution of
insulin and its precursors to fibrinogen and PAI-1 in a large population with
different states of glucose tolerance: the Insulin Resistance Atherosclerosis
Study (IRAS). Arterioscler Thromb Vasc Biol. 1999;19:562-567.
- Rao AK, Chouhan V, Chen X, Sun L, Boden G. Activation of the tissue factor
pathway of blood coagulation during prolonged hyperglycemia in young healthy
men. Diabetes. 1999;48:1156-1161.
- Balkau B, Bertrais S, Ducimetiere P, Eschwege E. Is there a glycemic
threshold for mortality risk? Diabetes Care. 1999;22:696-699.
- Balkau B, Shipley M, Jarrett RJ, et al. High blood glucose concentration
is a risk factor for mortality in middle-aged nondiabetic men: 20-year
follow-up in the Whitehall Study, the Paris Prospective Study, and the
Helsinki Policemen Study. Diabetes Care. 1998;21:360-367.
- Rodriguez BL, Lau N, Burchfiel CM, et al. Glucose intolerance and 23-year
risk of coronary heart disease and total mortality: the Honolulu Heart
Program. Diabetes Care. 1999;22:1262-1265.
- Jackson CA, Yudkin JS, Forrest RD. A comparison of the relationships of
the glucose tolerance test and the glycated haemoglobin assay with diabetic
vascular disease in the community: the Islington Diabetes Survey. Diabetes Res
Clin Pract. 1992;17:111-123.
- Kishimoto M, Yamasaki Y, Kubota M, et al. 1,5-Anhydro-D-glucitol evaluates
daily glycemic excursions in well-controlled NIDDM. Diabetes Care.
1995;18:1156-1159.
- Tamada JA, Garg S, Jovanovic L, Pitzer KR, Fermi S, Potts RO. Noninvasive
glucose monitoring: comprehensive clinical results. JAMA. 1999;282:1839-1844.
- Association AD. Standards of medical care for patients with diabetes
mellitus. Diabetes Care. 1999;22:S32-S41.
- Home PD. Rapid-acting insulin secretagogues: a clinical need? Exp Clin
Endocrinol Diabetes. 1999;107:S115-S119.
- Wolever TM, Chiasson JL, Josse RG, et al. Small weight loss on long-term
acarbose therapy with no change in dietary pattern or nutrient intake of
individuals with non-insulin-dependent diabetes. Int J Obes Relat Metab
Disord. 1997;21:756-763.
- Holt PR, Atillasoy E, Lindenbaum J, et al. Effects of acarbose on fecal
nutrients, colonic pH, and short-chain fatty acids and rectal proliferative
indices. Metabolism. 1996;45:1179-1187.
- Holman RR, Cull CA, Turner RC. A randomized double-blind trial of acarbose
in type 2 diabetes shows improved glycemic control over 3 years: UK
Prospective Diabetes Study 44. Diabetes Care. 1999;22:960-964.
- Wolever TM, Chiasson JL, Josse RG, et al. No relationship between
carbohydrate intake and effect of acarbose on HbA1c or gastrointestinal
symptoms in type 2 diabetic subjects consuming 30-60% of energy from
carbohydrate. Diabetes Care. 1998;21:1612-1618.
- Kado S, Murakami T, Aoki A, et al. Effect of acarbose on postprandial
lipid metabolism in type 2 diabetes mellitus. Diabetes Res Clin Pract.
1998;41:49-55.
- Hoffmann J, Spengler M. Efficacy of 24-week monotherapy with acarbose,
metformin, or placebo in dietary-treated NIDDM patients: the Essen-II Study.
Am J Med. 1997;103:483-490.
- Johnston PS, Lebovitz HE, Coniff RF, Simonson DC, Raskin P, Munera CL.
Advantages of alpha-glucosidase inhibition as monotherapy in elderly type 2
diabetic patients. J Clin Endocrinol Metab. 1998;83:1515-1522.
- Holman RR, Steemson J, Turner RC. Post-prandial glycaemic reduction by an
alpha-glucosidase inhibitor in type 2 diabetic patients with therapeutically
attained basal normoglycaemia. Diabetes Res. 1991;18:149-153.
- Rybka J, Goke B, Sissmann J. European Comparative Study of 2
alpha-glucosidase inhibitors, miglitol and acarbose. In: Program and abstracts
of the 59th Annual Meeting of the American Diabetes Association; June 18-22,
1999; San Diego, Calif; Abstract 433.
- Kawagishi T, Nishizawa Y, Taniwaki H, et al. Relationship between gastric
emptying and an alpha-glucosidase inhibitor effect on postprandial
hyperglycemia in NIDDM patients. Diabetes Care. 1997;20:1529-1532.
- Malaisse WJ. Mechanism of action of a new class of insulin secretagogues.
Exp Clin Endocrinol Diabetes. 1999;4:S140-S143.
- Bakkali-Nadi A, Malaisse-Lagae F, Malaisse WJ. Insulinotropic action of
meglitinide analogs: concentration-response relationship and nutrient
dependency. Diabetes Res. 1994;27:81-87.
- Marbury T, Huang WC, Strange P, Lebovitz H. Repaglinide versus glyburide:
a one-year comparison trial. Diabetes Res Clin Pract. 1999;43:155-166.
- Goldberg RB, Einhorn D, Lucas CP, et al. A randomized placebo-controlled
trial of repaglinide in the treatment of type 2 diabetes. Diabetes Care.
1998;21:1897-1903.
- Gomis R. Repaglinide as monotherapy in Type 2 diabetes. Exp Clin
Endocrinol Diabetes. 1999;107:S133-S135.
- Hu S, Wang S, Dunning BE. Tissue selectivity of antidiabetic agent
nateglinide: study on cardiovascular and beta-cell K(ATP) channels. J
Pharmacol Exp Ther. 1999;291:1372-1379.
- Hirschberg Y, McLeod J, Gareffa S, Spratt D. Pharmacodynamics and dose
response of nateglinide in type 2 diabetics. In: Program and abstracts of the
59th Annual Meeting of the American Diabetes Association; June 18-22, 1999;
San Diego, Calif; Abstract 430.
- Dimitriadis GD, Gerich JE. Importance of timing of preprandial
subcutaneous insulin administration in the management of diabetes mellitus.
Diabetes Care. 1983;6:374-377.
- Bolli GB, Di Marchi RD, Park GD, Pramming S, Koivisto VA. Insulin
analogues and their potential in the management of diabetes mellitus.
Diabetologia. 1999;42:1151-1167.
- Feinglos MN, Thacker CH, English J, Bethel MA, Lane JD. Modification of
postprandial hyperglycemia with insulin lispro improves glucose control in
patients with type 2 diabetes. Diabetes Care. 1997;20:1539-1542.
- Anderson JHJ, Brunelle RL, Keohane P, et al. Mealtime treatment with
insulin analog improves postprandial hyperglycemia and hypoglycemia in
patients with non-insulin-dependent diabetes mellitus: Multicenter Insulin
Lispro Study Group. Arch Intern Med. 1997;157:1249-1255.
- Lindholm A, McEwen J, Riis AP. Improved postprandial glycemic control with
insulin aspart. A randomized double-blind cross-over trial in type 1 diabetes.
Diabetes Care. 1999;22:801-805.
- Home PD, Lindholm A, Hylleberg B, Round P. Improved glycemic control with
insulin aspart: a multicenter randomized double-blind crossover trial in type
1 diabetic patients: UK Insulin Aspart Study Group. Diabetes Care.
1998;21:1904-1909.
- Scherbaum, WA. The role of amylin in the physiology of glycemic control.
Exp Clin Endocrinol Diabetes. 1998;106:97-102.
- Nyholm B, Orskov L, Hove KY, et al. The amylin analog pramlintide improves
glycemic control and reduces postprandial glucagon concentrations in patients
with type 1 diabetes mellitus. Metabolism. 1999;48:938-941.
- Thompson RG, Pearson L, Schoenfeld SL, Kolterman OG. Pramlintide, a
synthetic analog of human amylin, improves the metabolic profile of patients
with type 2 diabetes using insulin: the Pramlintide in Type 2 Diabetes Group.
Diabetes Care. 1998;21:987-993.
If you cannot register online and complete the activity, you may also receive
credit by mailing your completed Registration form, Post Test and Evaluation
Forms to:
|
Medical Education Collaborative
1800 Jackson
Street, Suite 200
Golden, CO 80401
Telephone: (303)
278-1900 |
Mealtime Glucose Excursions and Glycemic Control in Type 2
Diabetes CME
Register for CME Credit
| This information is needed for granting continuing
education credits and mailings regarding this website only. To receive CME
credits, or information concerning future activities, you must fill-in all
fields. There are no fees for participating and receiving CME credit for
this activity.
If you identify yourself with exactly the same Username and Password,
in all your submissions to us, we can associate the submissions, even if
you call in from different computers.
If you give us your email address, we can e-mail you notices about
future activities.
Contact Information
First Name:________________________
Last Name:_________________________
Email:______________________________
Social Security Number: ___________________________ (NOTE: US only)
Affiliation:_________________________________________
Address:______________________________________
City:________________________
State:______
Zip/Postal:____________
Country:__________________
Phone:_____________________
Fax:________________________
Professional Degrees
____ MD
____ DO
____ RN
____ PharmD
____
PhD
____ LPN
____ CNP
____ PhN
____ MSN
____ LVN
____
CCM
____ PA
____ NP
Credit Type for This Activity
____ Physician
____ Pharmacist
____
Nurse
Nurse or Pharmacist License: (required for
nurse or pharmacist CME credit)
State Licensed:_______
ID
Number:__________________
|
Mealtime Glucose Excursions and Glycemic Control in Type 2
Diabetes CME
Post Test
Click on the appropriate response. A test score of 70% or
greater is required for accreditation.
Mealtime Glucose Excursions and Glycemic Control in Type 2
Diabetes CME
Evaluation
| Scale: |
5 = Excellent |
4 = Good |
3 = Satisfactory |
2 = Fair |
1 = Poor |
All material on this website is
protected by copyright. Copyright ©
1994-2000 by Medscape Inc. All rights reserved. This website also contains
material copyrighted by 3rd parties. CME means Continuing Medical Education
credit is available. Medscape requires 3.x browsers or better from Netscape or Microsoft.